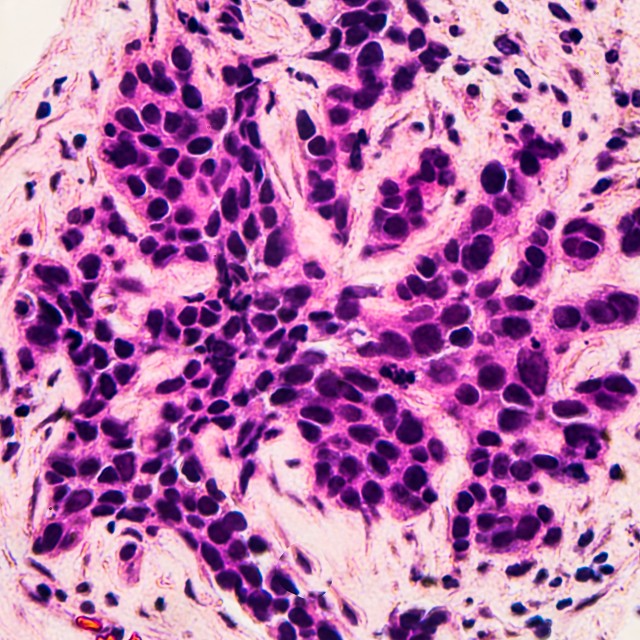
Image: Microscopic image (photomicrograph) of core biopsy for infiltrating (invasive) ductal carcinoma, detected by screening mammogram. H & E stain.
Our vision is a future where cancer is eliminated by total cure or managed by chronic treatment that enables a high quality of life. The mission of the Ben May Department is embodied in the motto of our founder, the late Nobel Prize winner Charles Huggins: "Discovery is our Business." In that spirit of discovery, our researchers are pushing the boundaries of understanding and challenging the assumptions that often impede progress. We believe the first step toward preventing or curing cancer is basic research on the intricacies of the human body and the molecular, cellular, and genetic events that lead to cancer. Advances in our fundamental understanding of cancer can then be translated into better methods of prevention and diagnosis.

Fitch Lectureship 2024
Monday, March 18, 2024 at 4:00 – 5:00 PM


Research
We believe that basic research on the molecular, cellular, and genetic events that contribute to carcinogenesis, metastasis and therapy resistance are the keys to making significant advances in cancer biology.
These advances have been and will continue to be translated into improved approaches to prevention, diagnosis and treatment. We benefit from interdisciplinary research opportunities and synergies across the University of Chicago.


